Part II of IV
The coughing is so violent, so thunder sharp, that Dr. Mike Welsh, hears the deep, wet claps through the room’s closed wooden door and well down the hallway.

The echo reverberates, the force of each choke conjuring a body in painful contraction. One of those rare moments when sound has a feel.
Inside, a young girl, 7 or 8, is fighting for each breath. That much is clear.
When she’s not coughing outright, her shallow breaths rattle, emitting a craggy low-pitched moan. And as she hawks, she brings up sputum, a custard-esque mucus. Smelling of rotting grapes, the phlegm is musty and acetone-tinged, like a decanter of red wine forgotten in a dark kitchen corner after a dinner party, left to turn into vinegar. Scientific name: Pseudomonas aeruginosa.
Welsh, a third-year med student on his pediatric rotation, watches how she’s recruiting every muscle to inhale: chest, diaphragm, neck, pecs even. And, hand on her back steadying a stethoscope, he feels the extreme effort in each exhale.
“Cystic fibrosis,” read the notes scribbled on her chart.
He’d learned about CF in his first two years of labs and lectures at the University of Iowa, sure, but textbook descriptions blunted the all-senses assault he was watching the disease wage on her small frame.
And, as he would learn, her spirit.
By now, the early 1970s, interventions had increased life expectancy for CF patients to just out of childhood. But the contours and origin of the illness remained unknown. It was more a syndrome, a collection of ailments, than a disease with a particular course.
And the therapies to ease symptoms were punishing.
Two hours, give or take, of lying upside down for “postural drainage” — treatments that rely on gravity to coax up gunk lodged in the lungs’ bottom reaches — as a caregiver rhythmically slaps your back. Imagine the ribs are a bottle, and the mucus is the ketchup desperately clinging to its sides, Welsh says.
All while breathing in mists through a nebulizer to open airways and loosen phlegm.
Even when she wasn’t stuck inside for her therapies, life as a sick child just wasn’t the same. With her lungs so blocked, she couldn’t run or jump or play like the other kids, and the fear of infection with a cold or a nasty bug clung to her loved ones like a shadow.
“We talk about how long someone lives, the survival time was this or that,” Welsh says. “But we don’t talk about all the suffering and the struggling during the time that you are alive, and that’s what people with CF had at that time.”
A child of John F. Kennedy’s moonshots, who sopped up the president’s speeches urging Americans to explore beyond their edges, Welsh always wanted an altruistic career. One where he, too, might plant a flag.
And, for Welsh, medical school was like Dorothy walking from sepia into technicolor. The right-left brain combination of bedside manner and biological detective needed in patient-facing medicine fit the natural philomath.
Bookish since his earliest days, Welsh learned compassion hanging from his mom’s apron strings. She was a farm wife, shouldering the home: raising four kids, growing their food, sewing their clothes, imparting wisdom rarely found in tests.
“My mom taught me empathy,” he says. “So many times, as a child, she would say, ‘How would you feel if …’”
She hoped to open him to new perspectives, yes, but new ways of thinking, too. His mother knew empathy fed curiosity like yeast to flour.
When Welsh learned about “physician-scientists” — medical doctors who saw patients sometimes and did bench research other times — he was fascinated by the permeability allowing the clinic to influence the lab and vice versa.
The path for the rest of his professional life seemed obvious after that, as though marked by flashing neon.
After the little girl’s exam, Welsh debriefs with the attending physician in the hall.
She’s probably not going to live into her teens, he tells Welsh. And if she does, well, she won’t make it out of her teens.
Doctors at the time “felt like their hands were tied,” says Bijal Trivedi, whose book “Breath from Salt” traces the rise of cystic fibrosis’ treatments. There were no good solutions, so many of them would end up telling parents to make their child comfortable.
“She was a just kid. She had her whole life ahead of her,” Welsh recalls. “How could that not impact you?”
The interaction, the way he felt in that hallway, propelled Welsh into pulmonary medicine and his research focus into chloride movement across the cells lining organs and cavities, which, by then, most understood as critical to cracking cystic fibrous.
He took a position in San Francisco after residency, where he saw the earliest cases of what would become the AIDS epidemic — another wave of healthy, young people dying despite every intervention. Totally different circumstances, and yet his helplessness, medicine’s helplessness, felt the same.
Watching parents become advocates, and even agitators, as they cradled their sons through their last moments showed him the power of a held hand. The first, but not final, time he’d see mothers and fathers mobilize love for therapeutic change.
Motivation is easy to find when you see so clearly how your research could impact human beings, Welsh says. Urgency, too, when no matter how hard you work, patients just keep dying.
“As a physician, there are certain people who are burned deep in your memory,” Welsh says. “That little girl I saw then, she’ll be with me the rest of my life.”
She became Welsh’s personal index case. The first patient — of many — whose story and memory he carried like ballast, steadying him against the gales.
The poet: A childhood of praying as science advances — slowly
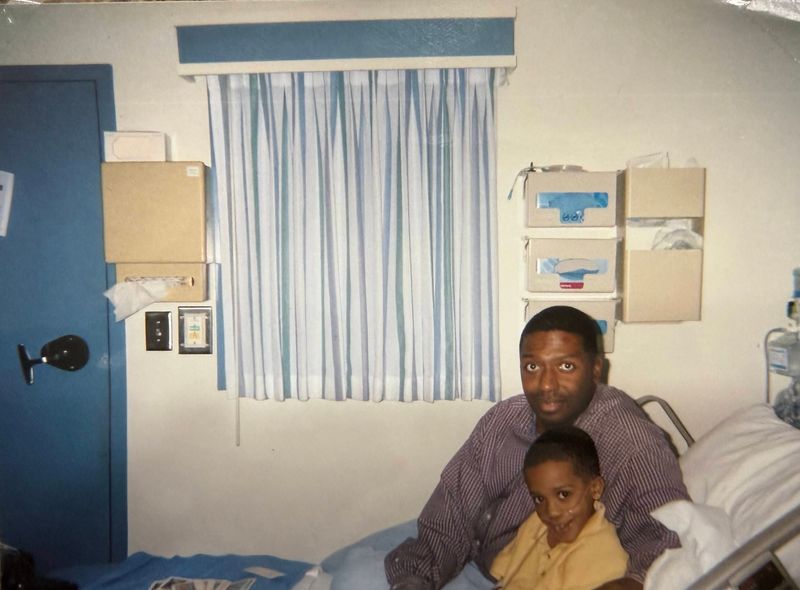
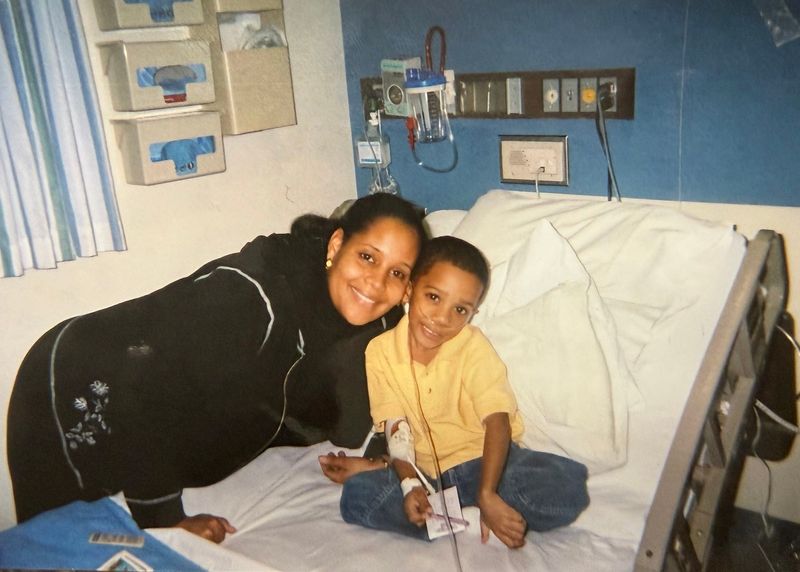
The doctors told William O’Neal II’s parents they would outlive their baby boy soon after his first cry.
He had a genetic disease, they believed, that would take him early. Incurable, doctors said.
They’d just become parents and now bereavement stood sentry to bliss. A staggering breakdown of the natural cycle right at the dawn of new life.
Sickle cell disease was the doctors first thought, a blood disorder disproportionately affecting Black people in which deformed cells become wedged in veins, leading to dangerous clots and extreme pain.
When tests returned a cystic fibrosis diagnosis around William’s three-month check-up, the horror was no less blunted, strokes substituted for suffocation.
By then, in 1999, William had a life expectancy of about 30 years. Maybe 40. At ab-so-lute best.
Already a rare disease, the O’Neals’ race made their cystic fibrosis story even more unique. The disease is passed from adult to child through an altered gene — like a predator in its cave, waiting for dusk — and Punnett Square genetics say both parents must be carriers to birth a baby with the disorder.
Even when both parents are carriers, there’s just a 25% chance of having a child with cystic fibrosis. And, in the U.S., only about 3% of CF patients are Black.
Which is to say: When the river was dealt, the cards didn’t lay their way. Nothing they could have done, doctors told them.
Fate, others said, a story written into their bodies generations ago.
The young parents left the hospital with a car seat, a baby blanket, a nebulizer for aerosol breathing treatments, a mechanical vest that shook violently to loosen mucus stuck in their baby’s lungs — the evolution of postural drainage — and a handful of pills. Crush them into a fine powder before spoon-feeding, doctors prescribed.
For O’Neal, illness was like saltwater to a shark. Sickness was his default, the framework around which all else hung. He didn’t get cancer or have an accident. He just was sick, now and forever.
His parents made sure their house was filled with enough love and stability to put up a fight against struggle. Research was developing, they told him, he just had to hold the line while they waited.
“My dad’s a pastor, so he was always like, ‘Miracles happen. Pray. Science is advancing,’” O’Neal says. “’There may be something that allows you to live longer. We just have to keep your lungs healthy enough until that happens.’”
They traveled as often as a preacher’s family could because of his mom’s job at Delta, and O’Neal looked forward to their trips, his stomach flipping with excitement as the plane climbed and climbed.
But cystic fibrosis was a tether, constantly yanking O’Neal back home for treatments. Pills. Aerosols. Vest. Half an hour at least. Twice a day. Every day. Repeat. Repeat. Repeat.
No overnights at friends’ houses. No summer camps. No movement, really, without thinking about his body.
So, isolation felt like a symptom of the disease. Just as much a manifestation of cystic fibrosis as the mucus coating his insides like blown-in insulation, making his body the perpetrator and victim of his slow smothering.
“When I was younger, I wanted to keep up with the other kids, run the mile in gym, you know?” he says. “I wanted to have a body that other people found desirable. I wanted to fall in love.”
“But I knew I had this thing that was different from everyone else,” he says. “Like, I was the odd one out.”
‘Dreaming away’ sickness in novels and movies and shows
O’Neal’s illness wasn’t a secret, but he stayed quiet about his struggles. Only a select few knew his private life of cystic fibrosis.
Classmates at his small Christian school outside Atlanta would have called him “nice,” he says. Teachers would have said “well-behaved.” “Unproblematic,” but to the point of banal feels apt for that version of himself, he says.
Mostly because shame was a symptom, too, he says. The mucus coughed into his hands, sometimes blood, both the reality of his lung muscles straining so much. The nausea that lasted three days when his pancreas flared from CF. The diabetes. The way all that meant his weight yo-yoed.
His anomalous body lacked decency.
Better to say clean and inconspicuous, like hotel artwork, which was fine for a preacher’s son.
For O’Neal, being part of a Black church in the South was an honor, a charge and a freight — all at once.
“In my small town in Georgia, it was like being the first family,” he says. “Everyone’s looking to you to decide what God is like. How you act is how God acts.”
“I was God’s son in the most real sense of the word.”
On Sunday, O’Neal’s father preached hope from the pulpit, stories of his family intertwined with morals and truths and parables. In O’Neal, congregants looked to see how God treated the sick. How He was clement.
And many saw his story as a catalyst for their own hope.
“I was the ‘church miracle’ in continuation,” O’Neal says. “But to be the spotlight of the continuous miracle that’s happening in front of your eyes, that’s a lot. Because then whenever you are sick, you’re like I have to get well.
“It was a lot of pressure not only to act well, but to act well through suffering.”
Twice a year, O’Neal would drop weight so precipitously and his breath would get so shallow, he’d need hospitalization. Two weeks of nearly round-the-clock treatments and observations.
So routine were these extended stays that some CF patients would keep go-bags, says Dr. David Stoltz, chief of the University of Iowa’s pulmonary division and one of Welsh’s mentees.
Often, he saw patients at the registration desk toting TVs or mini-refrigerators or the entire contents of their bathroom cabinets — as though checking into a dorm, not an ICU ward. They knew they’d be there for a while.
In his hospital room, O’Neal was alone most of the time. His family visited as much as possible: mom, dad and younger sister, 2½ years his junior. Cards flowed in from his father’s congregation, adding individualization to the sterile sameness of medicine.
And in the quiet, O’Neal began an education, parallel to the one he was missing at school.
“I was watching movies, and I was reading books and I was learning to write,” he says. “It was just literature, literature, literature.”
An instruction in life from C.S. Lewis. And paperback thrillers. And coming of age novels. And rom-coms.
He started developing his own stories, forming poems and plays.
O’Neal wrote about delinquents; characters whose exploits, their power, induced a creeping tingle on the back of your neck. A What Are They Going To Do Next kind of anxiety … or arousal, depending on which way you stared into the prism.
They had flings because he couldn’t. They imbibed because he couldn’t. They walked through portals to new worlds with heathens and heroes because his reality was all regional healthcare systems and make-up quizzes and God’s mercy.
“I was always trying to find ways to move past the limits of my body, and art is a way to do that,” O’Neal says. “I read a novel, and I can live three lives that I haven’t actually lived. I can write a novel and then write it over again.”
Having a chronic illness traps your soul in the body, O’Neal says. Imagination freed his spirit from his form, creating avatars far away from this one existence.
“My childhood was a lot of dreaming,” he says. “Dreaming away from my life that was just full of medications and illness.”
Because hope seemed wingless.
The turning point: A gene, a protein and the high of discovery
Welsh huddled over the lab’s fax machine as it screeched and squealed to life. In 1989, faxes ruled communication and this particular transmission — still hot from printing — had the power to change the direction of his research.
And his patients’ lives.
He’d received a call from the CF Foundation that an important update was coming in over the wire, and he had an idea of what it was: They’d found the gene that was defective in CF.
In the 1980s, gene mapping, or locating disease-causing genes, was gaining significant scientific traction. The mid-century advancements in DNA sequencing and genetic linkage had spurred a race to find the CF gene — which CF parents funded heartily hoping it would unlock the disease’s secrets.
A rare condition, effecting about 40,000 people in the U.S., parents understood CF was not an attractive area for research. Other diseases would simply have more impact. Cancer, for example. Heart disease.
They knew if they wanted the top minds, they were going to have to incentivize study.
So, since the Cystic Fibrosis Foundation’s charter in 1955, leadership pledged to invest as much money as possible in science. One longtime president’s mantra became: Money buys research, research buys cures.
They created a board of scientists that approved studies and experiments, which was “really, really novel,” Trivedi says. But parents were still the driving force, and they expected research to be completed collaboratively. Across universities. Across silos. One community.
Instead of ostentatious galas and publicity campaigns, they funded care centers where doctors could collect information about the disease. And then a registry that would allow scientists easy access to that data.
And then clinical trials. And the genetic research that led to Welsh’s fax that day in 1989.
But while some looked for the gene, Welsh studied the structure and inner workings of defective CF cells using salt as a guide star.
After San Francisco, he moved to Houston, where scientists were analyzing chloride movement at a more advanced level. He worked with a mentor who focused on the intestines and, specifically, how the bowels regulated salt and water.
At the end of the day, Welsh collected animal lungs from other labs and worked on his own experiments, using tools and theories he picked up during his work with the gut.
One night, while measuring the electrical properties of the trachea and the flow of chloride across the cells, he had surprising results, findings he’d never seen previously.
He checked the journals for information. Nothing like this had already been published.
“I went home at 3 or 4 in the morning, and I couldn’t sleep. I was thinking about my experiment,” Welsh says.
“I thought about it all the time: when I was eating, when I was at the grocery store, when I was at a movie. I just couldn’t think about anything else except my experiments.”
When he finally figured out why he was getting such a unique set of outcomes, the high was physical, like the all-body prickling sensation when a blast of cold air conditioning spikes sweaty skin, crystalizing the beads.
His first taste of true discovery.
“Perhaps it’s like an explorer from a long time ago discovering a continent,” he says.
Everyone else looked and only saw endless sea. But there was, he knew, something on the horizon.
Welsh made his way back to Iowa in 1981, and in the years that followed, his research pinpointed that chloride wasn’t passing through cell membranes in people with cystic fibrosis. Instead, chloride seemed to get caught inside the cell, which disrupted the flow of water into the airways. And without liquid, the naturally fluent mucus inside the lungs became gummy and hard to clear.
But Welsh still didn’t know why. He didn’t know how the chloride was being penned in or what was causing the blockage.
How can this be? he thought as he kept investigating, following new lines of inquiry. Experiment after experiment.
Curiosity had driven him this far, but he’d need a breakthrough, a new discovery, to make real headway.
And then the fax started beeping.
There are many ways to fix a car with a broken engine … but what about a gene?
The two main research tracks of the 1980s, Welsh’s and others’ gene-mapping, took divergent views on the same problem, Welsh told the Register at the time.
“It’s like trying to find out why a car engine won’t start,” he said. “One approach is to tear the motor apart to find the fault. The other is to look at blueprints to compare cars that do and don’t work.
“They found there was a line missing in the blueprint.”
Indeed, the fax Welsh was waiting to grab off the tray announced that after studying hundreds of thousands of letters of code, two scientists, Francis Collins and Lap-Chee Tsui, and their teams had discovered three missing letters in the DNA of a group of people with CF.
The deletion altered the production of a protein, which would become known as CFTR, a merciful shorthand for cystic fibrosis transmembrane conductance regulator.
And that broken protein set off the biological chain reaction Dorothy Anderson, the brilliant if unconventional female scientist to first identify CF, had seen decades and decades ago in the basement of Babies Hospital in New York City.
“That was a turning point,” Welsh says. “But that opened up so many questions.”
There had been some success with gene therapy in other diseases, and the goal of finding the gene was to replicate that in CF. Now, the scientists and the parents that funded them hoped they could use innocuous viruses as trojan horses, delivering cells with healthy genes that would kick-start the protein and correct the problem.
“Take a cold virus, swap out the DNA that the cold virus is carrying, put in the CFTR DNA, and give that to a person,” Welsh explains.
Some of these early attempts at gene therapy were partly successful. In fact, Welsh was among the first to prove that in cultures in the lab, CF cells accepted the healthy DNA and started producing normal CFTR. But those wins turned out to be smokescreens when human trials started: The lung had evolved over millions of years to keep out bacteria and viruses — even good ones.
The gene was always just one of the scientific nesting dolls toward a cure, Welsh knew, so what would it look like if he focused on the next matryoshka?
“His power of observation is as strong in the research setting as it was in the clinical setting,” says Stoltz, who trained with Welsh.
“We could be looking at a CAT scan and somebody else may say, ‘That’s pneumonia.’ But he would see something nuanced on the CAT scan and say, ‘Why is it like that?’ Or, ‘Have you seen this before?’”
So, Welsh thought, instead of fixing the gene, what if he fixed what the gene made — the protein itself?
Welsh and his team would need to know a lot more about its structure and function to do so. The gene was just directions, after all, a section of DNA written in a four-letter language, but they were blind to its end product.
“You don’t know what those instructions do. Do they encode a car? Do they encode a spacecraft? Do they encode a toaster?” Welsh says. “What do those instructions build inside of a cell?”
For a year, Welsh and his collaborators struggled to reproduce the protein in their lab-grown cells, which they’d need to do to study its purpose and mechanics.
The bacterium researchers used as an incubator before DNA was placed into a cell kept mutating the strand’s sequence for some technically complex reasons, Welsh says.
So, scientists had to make tiny adjustments to the gene’s DNA until it could manufacture the protein correctly. Flinty work that felt impossible some days, like grabbing a cold front in your fist to hold back falling temperatures.
Then one Sunday, a car came rambling up Welsh’s long driveway in the country, out in the Amish part of Iowa’s Johnson County. One of his researchers held an X-ray film in her hand like a Broadway producer with an opening night review.
A tiny radioactive mark showed they’d finally succeeded in recreating the protein. A hit.
And the next day, Welsh asked for a sabbatical.
He wasn’t leaving the university, nor his research.
But he needed to channel all his energy into this protein, and only this protein.
His mind was already racing, like it had back in Houston: What’s next? How do we get there? Will it help my patients?
The patients he carried — some now spirits over his shoulder — demanded his focus.
Courtney Crowder is the Register’s Iowa Columnist and a senior writer. Please share stories and tips at ccrowder@dmreg.com or 515-284-8360.
The Miracle of Breath series continues
This is Part 2 of a four-part series. Start from the beginning here or continue reading Part 3.
Future parts will describe how Dr. Mike Welsh’s meticulous research led to an easily overlooked breakthrough, why a Eureka moment facilitated groundbreaking chemistry and what waiting on death does to the mind and spirit — until one phone call overrides fate.
The Miracle of Breath series and “Giving Back Breath,” its companion documentary, trace the story of how Dr. Mike Welsh’s meticulous experimentation led to a breakthrough that transformed cystic fibrosis from a lethal diagnosis into a manageable condition.
Told through the intimate, unbelievable night Welsh met one of the people his work saved in an Iowa City bar, the 20-minute documentary is a co-production of the science-focused production company HHMI Tangled Bank Studios, USA TODAY Co. and the Des Moines Register.
Watch the film online and then dive deeper into the series.
Watch the ‘Giving Back Breath’ documentary free on YouTube
Influential Iowans
This is the last of the Des Moines Register’s series on Influential Iowans. Through the end of 2025 and the start of 2026, the Register has publish profiles of people who have shaped our state and the country beyond. These people influence our politics, food, sports, communities, music, city development and arts, and health. They were chosen by Register staff ― and each is fascinating on their own. Together, they show the rich fabric of Iowa’s influence.
— Rachel E. Stassen-Berger, Des Moines Register executive editor
This article originally appeared on Des Moines Register: An Iowa scientist chases a medical mystery as patients cling to breath
Reporting by Courtney Crowder, Des Moines Register / Des Moines Register
USA TODAY Network via Reuters Connect